Peform a differential expression analysis.
Source:R/fdge.R, R/fdge-compare.R, R/fdge-interact.R, and 1 more
fdge.RdUse flm_def() to define the design matrix and contrast to test and
pass the FacileLinearModelDefinition object returned from that to fdge()
to run the desired differential testing framework (dictated by the method
parameter) over the data. flm_def accepts a
fdge(
x,
assay_name = NULL,
method = NULL,
features = NULL,
filter = "default",
with_sample_weights = FALSE,
treat_lfc = NULL,
...,
verbose = FALSE
)
# S3 method for FacileAnovaModelDefinition
fdge(
x,
assay_name = NULL,
method = NULL,
features = NULL,
filter = "default",
with_sample_weights = FALSE,
...,
verbose = FALSE
)
# S3 method for FacileTtestDGEModelDefinition
fdge(
x,
assay_name = NULL,
method = NULL,
features = NULL,
filter = "default",
with_sample_weights = FALSE,
treat_lfc = NULL,
...,
verbose = FALSE
)
# S3 method for FacileLinearModelDefinition
fdge(
x,
assay_name = NULL,
method = NULL,
features = NULL,
filter = "default",
with_sample_weights = FALSE,
treat_lfc = NULL,
weights = NULL,
with_box = FALSE,
...,
biocbox = NULL,
trend.eBayes = FALSE,
robust.eBayes = FALSE,
verbose = FALSE
)
# S3 method for FacileTtestAnalysisResult
compare(x, y, treat_lfc = param(x, "treat_lfc"), rerun = TRUE, ...)
# S3 method for FacileTtestComparisonAnalysisResult
tidy(
x,
max_padj = 0.1,
min_logFC = NULL,
labels = NULL,
...,
max_padj_x = max_padj,
max_padj_y = max_padj,
min_logFC_x = min_logFC,
min_logFC_y = min_logFC
)
# S3 method for FacileTtestAnalysisResult
report(
x,
type = c("dge", "features"),
ntop = 200,
max_padj = 0.1,
min_logFC = 1,
features = NULL,
highlight = NULL,
round_digits = 3,
event_source = "A",
webgl = TRUE,
caption = NULL,
...
)
# S3 method for FacileTtestComparisonAnalysisResult
ffsea(
x,
fsets,
methods = NULL,
type = c("interaction", "quadrants"),
features = NULL,
min_logFC = param(x, "treat_lfc"),
max_padj = 0.1,
rank_by = "logFC",
signed = TRUE,
biased_by = NULL,
...,
rank_order = "ranked",
group_by = "direction",
select_by = "significant"
)Arguments
- x
a data source
- assay_name
the name of the assay that holds the measurements for test. Defaults to
default_assay(x).- method
The differential testing framework to use over the data. The valid choices are defined by the type of assay
assay_nameis. Refer to the Differential Expression Testing Methods section for more details.- features
Explicitly request the features to test. If this is provided, then the
filtercriteria (to remove low abundance features, for instance) is skipped. By default this isNULL- filter, with_sample_weights
Passed into
biocbox()to determine which features (genes) are removed from the dataset for testing, as well as if to uselimma::arrayWeights()orlimma::voomWithQualityWeights()(where appropriate) when testing (default is not to).- ...
passed down into inner methods, such as
biocboxto tweak filtering criteria, for instance- weights
a
sample_id,feature_id,weighttable of observation weights to use whenmethod == "limma".- rerun
When comparing two results, the features analyzed in each may differ, making comparisons between the two objects sparse, at times. When
rerun = TRUE(default), the original linear models are rerun with their features set tounion(features(x), features(y)).
Differential Expression Testing Methods
The appropriate statistical framework to use for differential expression
testing is defined by the type of data that is recorded in the assay
assay_name, ie. assay_info(x, assay_name)$assay_type.
The fdge_methods() function returns a tibble of appropriate
assay_type -> dge_method associations. The first entry for each
dge_method is the default method used if one isn't provided by the
caller.
The available methods are:
"voom": For count data, useslimma::voomWithQualityWeights()whenwith_sample_weights = TRUE."edgeR-qlf": The edgeR quasi-likelihood method, for count data."limma-trend": Usable for log-transformed data that "looks like" it came from count data, or where there is a "trend" of the variance with the mean, useslimma::arrayWeights()whenwith_sample_weights = TRUE."limma": Straightup limma, this expects log2-normal like data, with (largely) no trend of the variance to the mean worth modeling. Useslimma::arrayWeights()whenwith_sample_weights = TRUE
Feature Filtering Strategy
You will almost always want to filter out lowly abundant features before
performing differential expression analysis. You can either do this by
explicitly requesting which features to test via the features parameter,
or by setting filter = "default.
When filter == "default", the filtering strategy is largely based on the
logic found in edgeR::filterByExpr().
When fdge analysis is performed on count data, the filtering is precisely
executed using this function, using design(x) as the design parameter to
filterByExpr. You can modify the filtering behavior by passing any
named parameters found in the edgeR::filterByExpr() function down to it via
fdge's ... parameter (don't pass design, as this is already defined).
There are times when you want to tweak this behavior in ways that aren't
exactly supported by filterByExpr. You can pass in a "feature descriptor"
(a character vector of feature ids, or a data.frame with a "feature_id"
column) into the following parameters:
filter_universe: The features enumerated in this parameter will restrict the universe of features that can potentially be included in the downstream analysis. ThefilterByExpr()logic will happen downstream of this universe. The default value isNULL, which specifies the universe of features to be all of the ones measured using this assay.filter_require: ThefilterByExprlogic happens first on the universe of features as parameterized. All features enumerated here will be forcibly included in the analysis, irrespective of whether they would have passed the perscribed filter criteria or not. The defalut value for this argument isNULL, which means no genes are forcibly included in the analysis when they do not pass muster given the filtering criteria.
Comparing DGE Results (interaction effect)
It is often useful to compare the results of two t-tests, and for many experimental designs, this can be a an intuitive way to perform test an interaction effect.
The filtering strategy in the interaction model dictates that the union
of all features found in x are y are used in the test.
Statistics Tables
The stats table from the differential expression analysis can be retrieved
using the tidy() function. Depending upon the type of analysis run, the
exact columns of the returned table may differ.
Interaction Statistics
Calling tidy() on an interaction test result
(FacileTtestComparisonAnalysisResult), returns the statistics for the
interaction test itself (if it was performed), as well as the statistics
for the individual DGE results that were run vai compare(x, y) to get the
interaction results itself. The columns of statistics related to the
individual tests will be suffixed with *.x and *.y, respectively.
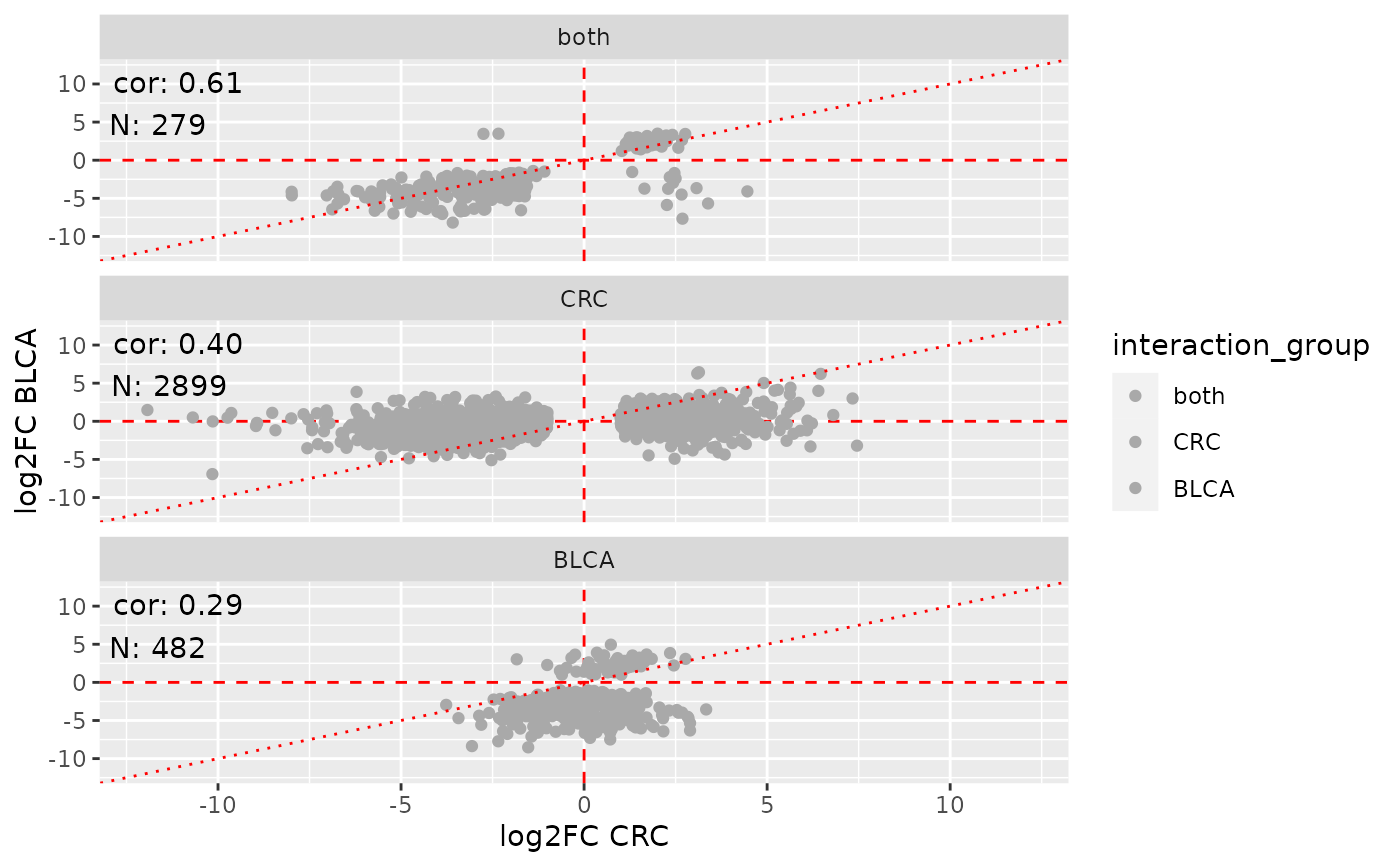
An interaction_group column will also be added to indicate what type of
statisticaly significance was found for each gene. The values in here can be:
"both": this gene was statistically significant in both tests"x": this gene was only significant in thexdge result"y": this gene was only significant in theydge result"none": was not statistically significant in either test result
The genes selected for significance from the input results x and y are
based on their padj and logFC values. The thresholds are tweaked by the
following parameters in the call to tidy(compare(x,y)):
max_padj_(x|y): if not specified, defaults to0.10min_logFC_(x|y): if not specified, we will take the value that was used in thetreat_lfcparameters toxandyif those tests were run against a threshold, otherwise defaults to1.
Interacting with results
The report function will create an htmlwidget which can be explored by
the analyst or dropped into an Rmarkdown report.
report(result, "dge", max_padj = 0.05, min_logFC = 1) will create a
side-by-side volcano and datatable for differential expression results.
Gene Set Enrichment Analysis
There are a few ways you may consider running a gene set analysis over an interaction analysis.
On the statistics of the interaction itself; or
On the statistics of the different "quadrants" the features are binned into that are found in the
sigclasscolumns of the tidied result table, ie.tidy.FacileTtestComparisonAnalysisResult(); orBoth.
Note that an analysis on (2) only lends itself to an overrepresentation
analysis, ie. methods = "ora".
Examples
efds <- FacileData::exampleFacileDataSet()
samples <- efds %>%
FacileData::filter_samples(indication == "BLCA") %>%
dplyr::mutate(something = sample(c("a", "b"), nrow(.), replace = TRUE))
mdef <- flm_def(samples, covariate = "sample_type",
numer = "tumor", denom = "normal",
batch = "sex")
dge <- fdge(mdef, method = "voom")
if (interactive()) {
viz(dge)
viz(dge, "146909")
shine(dge)
}
dge.stats <- tidy(dge)
dge.sig <- signature(dge)
stage.anova <- samples %>%
flm_def(covariate = "stage", batch = "sex") %>%
fdge(method = "voom")
anova.sig <- signature(stage.anova)
# Comparing two T-test results ----------------------------------------------
# Let's compare the tumor vs normal DGE results in CRC vs BLCA
efds <- FacileData::exampleFacileDataSet()
dge.crc <- FacileData::filter_samples(efds, indication == "CRC") %>%
flm_def("sample_type", "tumor", "normal", "sex") %>%
fdge()
dge.blca <- FacileData::filter_samples(efds, indication == "BLCA") %>%
flm_def("sample_type", "tumor", "normal", "sex") %>%
fdge()
dge.comp <- compare(dge.crc, dge.blca)
comp.hi <- tidy(dge.comp) %>%
dplyr::group_by(interaction_group) %>%
dplyr::slice(1:3) %>%
dplyr::ungroup()
# Static visualization generates the main "4-way" plot, as well as the
# facets for each category.
sviz <- viz(dge.comp, labels = c(x = "CRC", y = "BLCA"),
subtitle = "Tumor vs normal comparisons across indications",
highlight = comp.hi)
# highlight some of them
s.hi <- sviz$input_data %>%
dplyr::group_by(interaction_group) %>%
dplyr::slice(1:3) %>%
dplyr::ungroup()
if (requireNamespace("patchwork")) {
patchwork::wrap_plots(
sviz$plot + ggplot2::theme(legend.position = "bottom"),
sviz$plot_facets + ggplot2::theme(legend.position = "none"),
nrow = 1)
viz(dge.comp, labels = c(x = "CRC", y = "BLCA"),
color_quadrant = "darkgrey")$plot_facets
}
#> Loading required namespace: patchwork